Methods for estimating the diversity of eukaryotic protists
Back to main indexAn estimate of biodiversity must begin with a local analysis of the different biotopes. In the case of microbial life, these biotopes will be very numerous. Indeed, the small size and metabolic specificity of microorganisms sometimes only allows them to occupy very limited and surprising niches: the microbial content of the digestive tract of termites varies according to the species and the region of the tract, the top and bottom of leaves host different microbiota - just like the inside of the leaf; in humans the different parts of our body are colonized by specific prokaryotic and eukaryotic microorganisms; the joints dishwashers or bathtubs are colonized by a defined fungal flora, etc. The analysis of different biotopes is therefore far from over and often the study of new ecological niches makes it possible to highlight a large number of new species. For example, by exploring the diversity of yeasts present in the gut of beetles, approximately 200 new species were described in 2005. This represented an increase of approximately 30% of the known species of yeast! Analysis of the rate of discovery of new species suggested that about as many new yeasts remained to be discovered in the same biotope…
Several methods exist to determine local diversity. The oldest and most direct is to look at the morphology and behavior of harvested specimens, most often under the microscope. This method provides a list of species based on morphological criteria. It requires many observations and vast skills, especially if one is interested in the diversity of all biological groups! It also requires a good knowledge of the complete life cycle of organisms, because they can appear in very different forms depending on the conditions or developmental stage. Likewise, biotopes undergo changes which lead to defined successions, because species can occupy a biotope and transform it, thus making it usable for successive species in the same biotope. The term to make usable does not necessarily mean that the following species find an “improved” environment, but on the contrary often a poorer environment on which they are more competitive. This succession can be true, that is to say that species are present in turn in the biotope, or apparent, meaning that the species are always present but are only detectable to certain moments in time. For example, many fungi seem to grow successively in the excrements of herbivores, the coprophilic fungi (Figure 68). This succession, which recapitulates the evolution, is reproducible and takes a few weeks. However, the fungi are all inoculated into this biotope in the form of spores when the herbivore eats the plants. In fact, their spores often germinate specifically as they pass through the digestive tract. In some cases, these successions extend over several years or even tens or hundreds of years. For example, in a Swiss forest, an area of 1,500 m2 has been analyzed every week for 21 years. 71,222 carpophores belonging to 408 different species were recorded. Only eight species have been found each year and new species are found each year. 19 species have fruited for the first time in the last harvest year!
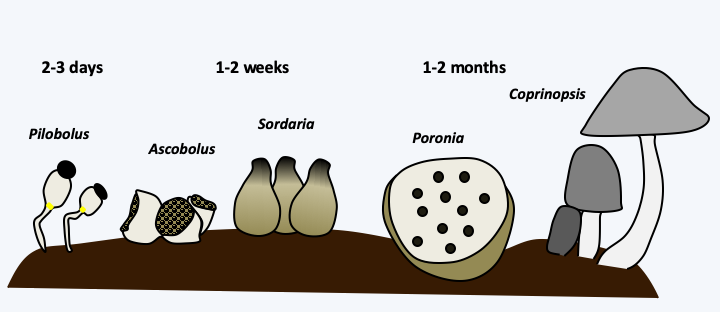
Figure 68.
Succession of coprophilic fungi. Over 2,000 species of fungi live on herbivorous droppings. They follow one another in a relatively defined way. The succession begins with the appearance after a few days of non-dikarya fungi of the Mucoromycotina type such as Pilobous, then of Ascomycota Pezizomycetes such as Ascobolus or Saccobolus, a few days later of Sordariomycetes such as Sordaria and Podospora, followed a few weeks later by Poronia. The succession often ends with Basidiomycota such as Coprinopsis, Cyathus or Stropharia. All were ingested at the same time by the herbivore and their mycelium is present since the deposit of the excrement. The kinetics of onset depend on the growth rate of different species, their ability to degrade plant residues and antagonisms that allow late species to eliminate early species.A second method consists in the cultivation, either on Petri dishes containing the appropriate medium or in liquid cultures, using different types of media (Figure 69). This technique obviously comes up against the fact that many species cannot be cultivated. Conditions may be unsuitable, as many species require special foods. Others have states of dormancy that require special conditions to get out. They can be fragile and die at the time of harvest or live in a symbiosis, etc. It is estimated that only a few percent of the cells present in a sample give rise to a culture. Nevertheless, recent advances have allowed the cultivation of more and more previously recalcitrant organisms. It is often enough just to be patient, as many microorganisms have long generation times. Another problem is that the species harvested are in fact those that grow well in culture, while they are not necessarily the majority in nature. Molecular studies on the fungal diversity of the rhizosphere of wheat carried out as early as 1995 clearly illustrate this difficulty. Two pairs of primers were used to specifically amplify DNA derived from the 18S rRNA gene from DNA extracted directly from the rhizosphere; the two pairs are necessary to obtain the widest possible range of fungi while excluding other eukaryotes. Of 61 analyzed clones, 24 species belonging to different genera of fungi were identified. However, this method failed to detect species of the genus Fusarium which are nevertheless easily cultivable from the same soils, suggesting that Fusarium could in fact be in the minority in terms of quantity. Other experiments show, for example, that isolating DNA from a soil sample and culturing the same sample can identify 51 and 67 species, respectively. Only one species is identified by both methods! Despite its shortcomings, the cultivation method allows for a more in-depth study of the biology of the species. A good culture currently teaches even more about the biology of an organism than a DNA sequence. It is therefore still widely used. It is often coupled with the obtaining of sequences which will make it possible to analyze the phylogenetic position of the species harvested. If the sample is of high interest, a complete genome sequence can be obtained, as new sequencing technologies are now relatively inexpensive. Otherwise, only one diagnostic region of the genome is examined. This is also what is done if many samples have to be analyzed. The sequenced region, called “barcode”, depends on the species. In the case of Eumycota fungi, the most used barcode is the intergenic sequence located between the 28S and 18S rDNA, called ITS for “intergenic transcribed spacer”. In algae and protozoa, the recommended barcodes are D2 and D3 parts of the ribosomal large subunit gene and the mitochondrial cox1 gene encoding a cytochrome oxidase subunit.
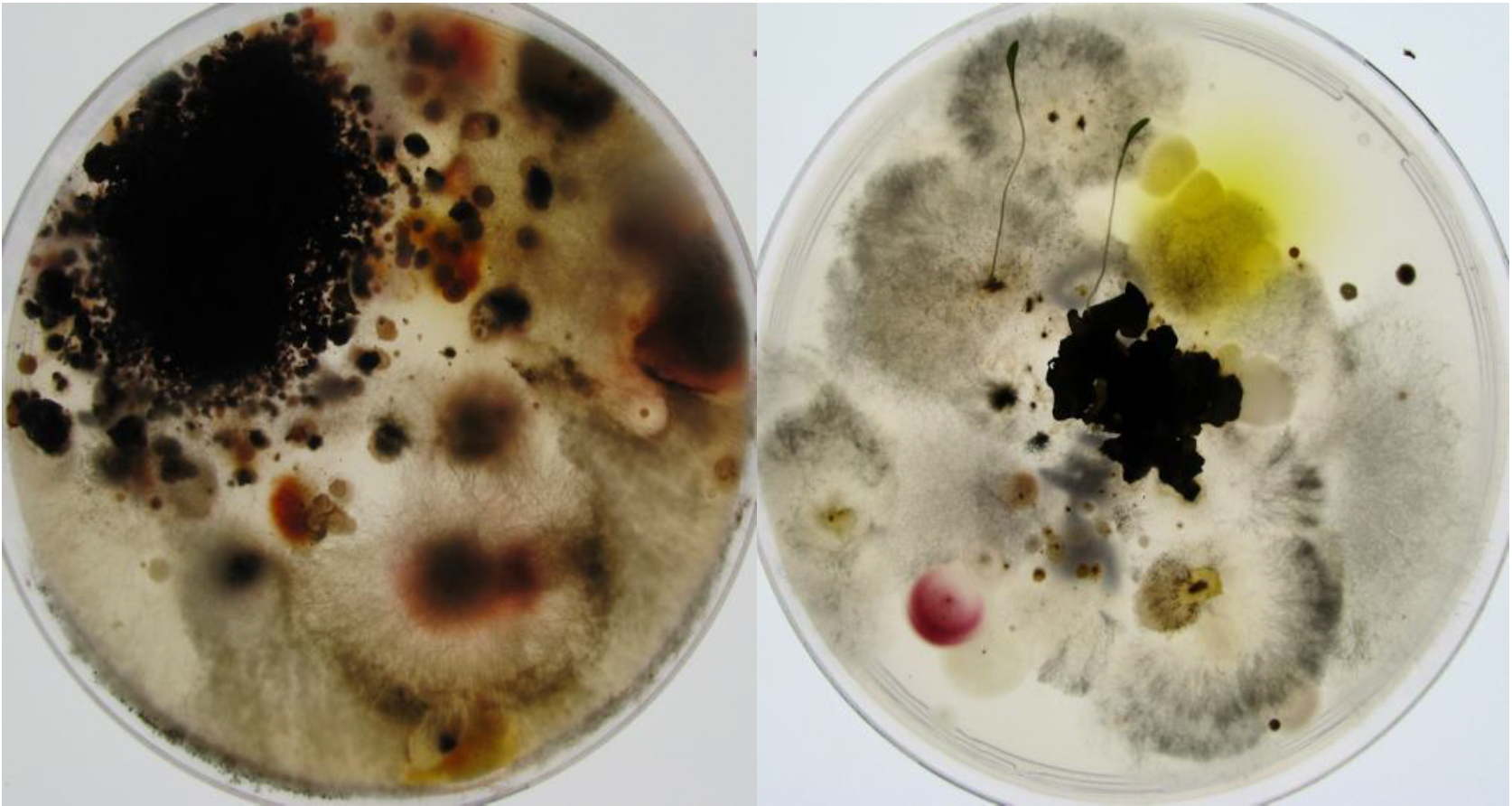
Figure 69.
Examples of cultivation directly from a soil sample. A few milligrams of soil from a meadow were deposited on two different media allowing fungal growth. Both media contain antibiotics to limit bacterial growth. Each of the media (rich on the right, poor on the left) gives different species of fungi. Two seeds have germinated on the middle of the right.Advances in high throughput sequencing methods not only make it possible to obtain the genome sequences of many organisms at low cost, but also to exhaustively investigate the DNA, and therefore the species present, in a given biotope. It suffices to extract DNA directly from samples of media: soil, mud, intestinal contents, etc. This DNA is then used during a PCR amplification experiment with primers derived from genes encoding RNAs or proteins conserved during evolution. The most frequently used genes in the case of eukaryotic microbes are the ones encoding 18S and 28S rRNAs. These RNAs comprise different regions which evolve more or less quickly. By using ad hoc primers, it is therefore possible to amplify different regions, specific to more or less important biological groups. The amplification products are cloned and the various clones obtained are sequenced at high throughput. The comparison of the resulting sequences with databases makes it possible to identify the species present, and the number of clones obtained having the same sequence is an estimate of the size of the different populations (Figure 70). This technique, called metagenomics, has been used for a long time; the analysis of the fungal flora around the roots described above is an example. But, it has only recently shown its full power with the drop in sequencing costs. Now millions of sequences can be generated for a few hundred euros, allowing for sampling of minority species as well. The results show that most often the sequences thus obtained are not in the databases and come from species still unknown or already cultivated but not studied, confirming that a large part of the biodiversity of eukaryotic microbes is still unknown. They also confirm that the majority of species are not necessarily those which can be cultivated or which are easily observed.
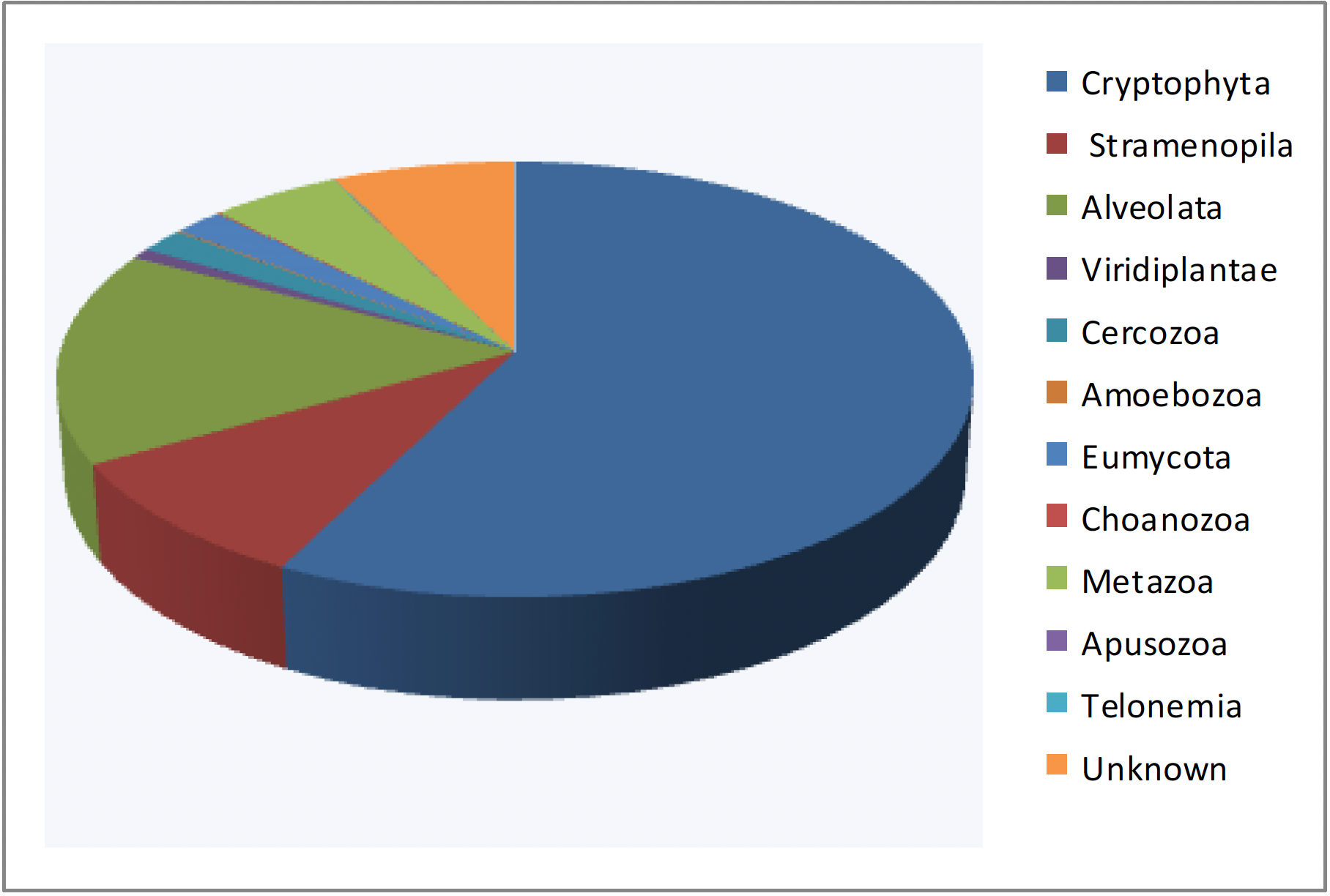
Figure 70.
Typical result of a metabarcoding analysis. Sequence analysis of 18S RNA genes amplified from DNA extracted from sediments from Lake Finsevatn in Norway shows that the majority eukaryotic group is Cryptophyta. Most of the sequences obtained belong to unknown species, but having clear affinities with one of the major groups of eukaryotes. However, there are still around 8% of species without any affinity (unknown).Back to chapter index