Molecular phylogenies - successes and setbacks
Back to main indexThe advent of gene and protein sequence studies has profoundly changed phylogeny. Indeed, the comparison of RNA and protein sequences that are conserved in eukaryotes, such as translation elongation factors, hsp, tubulins, etc., in theory makes it possible to trace the evolutionary history of eukaryotes according to the precept that the more similar the sequences are, the more closely related the organisms are, and vice versa. This method has been very successful and has indeed resulted in a much better natural phylogenetic classification than previous ones. To do this, there are several methods of calculating distance: parsimony, neighbor joining, maximum likelihood, etc., the analysis of which is outside the scope of this book. However, when one reads the very abundant literature on the subject, one is struck by the contradictions that one can find, according to the different analszes. This is especially true in the case of relationships between large clades of protists, but also large clades of animals and plants! Where can the discrepancies come from? Several reasons can be cited:
- the use of too short sequences, leading to alignments where chance has a very large effect. In this case, depending on the distance calculation methods chosen, the results can be very different, especially if the sequences are very divergent. Currently, this problem is often solved by using the sequences of several genes, or even complete genomes.
- the number and range of species used are too small for the reconstruction we are trying to achieve. In this case, the sequences are very divergent and the phylogenetic signal too weak. It is then necessary to increase the number of species, leading to an increase in the computing time. Increasing the computing power of often solves the problem, although comparison algorithms increase computing time exponentially with the number of sequences compared.
- the use of genes that change too fast or too slowly. For example, if a gene evolves very quickly, the many changes that have occurred at the same position, some of which may cancel out the previous ones, lead to an underestimation of the distance between the species studied. This is often the case with rRNAs on which a large fraction of phylogenetic trees are based. Another possibility is that a given gene changes at different rates in different lineages. This leads to branches of different lengths in the trees. In this case, it has been shown that the fastest evolving species with the longest branches may incorrectly cluster at the base of the tree. This phenomenon is called “long-branch attraction”. It is highly probable that the grouping of eukaryotes without mitochondria at the base of the tree, as observed in early phylogenies, came from this phenomenon. Currently, methods correcting the possibility of multiple changes and “good genes” evolving at the right speed are used to overcome the problem. These genes are different from one phylogenetic group to another.
In addition to these problems associated with using too little information to establish phylogenies, two other mechanisms are sometimes involved in obtaining false phylogenies. A first mechanism is the horizontal transfer of sequences between different species. It seems that this process had a less important role in eukaryotes than in prokaryotes. However, many cases of horizontal transfers have been demonstrated either from prokaryotes to eukaryotes, or between eukaryotes. While this mechanism is quite easily explained in the case of phagotrophic organisms, because the DNA of the prey can escape from the digestion vesicle and join the nucleus, or photosynthetic organisms because the plastid is a source of prokaryotic genes, it is more mysterious in fungi. Nevertheless, many cases of transfers between true and false fungi have been highlighted. For the time being, the involvement of viruses as a vector remains can be documented. The frequency of this phenomenon could be underestimated, in particular in the case of repetitive transfers, the most emblematic case of which is probably that of the genes encoding the translation factor eEF1A. This essential protein is involved in translation. It is widely used to establish molecular phylogenies because this highly conserved factor makes it possible to detect kinship relationships between distant organisms. The major isoform of this “eEF1A” gene has been repeatedly replaced by the “EF-like” or EFL isoform (Figure 77). The frequency of transfers is such that they probably took place via a vector, possibly a virus. The use in comparisons of genes derived from two copies of a duplication or paralogs, and which therefore do not come from a common ancestor such as orthologs (Figure 78), can also lead to false phylogenies. The phenomena of so-called segmental duplications, diploidizations or hybridizations which lead to the creation of paralogous copies are very frequent. When coupled with deletions, artefacts appear. Initiated with the use of ribosomal RNAs, then very conserved proteins such as translation elongation factors eEF1A and eEF2, cytoskeletal proteins (tubulins, actin) or RNA polymerase subunits, molecular phylogenies continue to resolve the phylogenetic position of many eukaryotic protists, especially when using concatenated sequences of multiple genes. It seems that 20 carefully chosen genes are often sufficient to obtain the correct phylogeny, but with the advancement of sequencing methods it often becomes faster to obtain the complete sequence of genomes. However, not all problems are solved by these methods. In particular when rapid evolutionary radiation has occurred and/or the groups have diverged for a very long time, which is the case for eukaryotes, the phylogenetic signal is often too weak and relationships between the different lineages are then impossible to determine on the sole basis of the sequences. It is then possible to search for molecular signatures. These are rare events, and if shared, indicate a kinship relationship, even though the organisms are very different. These are for example the fusion of two coding sequences into one, the deletions or insertions (= indels) of precise sequences, the presence of an enzyme or an original metabolic pathway (but beware of horizontal transfers!). For example, the fusion of the couple of enzymes involved in the synthesis of thimidine, dihydrofolate reductase and thymidilate synthase, into a single polypeptide in the eukaryotes Excavata and Diaphoretickes while in all other organisms, Bacteria, Archea and Amorphean eukaryotes, the enzymes are encoded by two different polypeptides, suggested that Excavata and Diaphoretickes are related. The eukaryotic root would then be placed between Amorphea on the one hand and “Excavata + Diaphoretickes” on the other. However, it is clear that the current fault in the development of most phylogenies is the exclusive use of molecular data, sequences or signatures. Indeed, the evolution of eukaryotes is very complex involving many symbioses, convergent evolution also occurring at the molecular level and recurrent horizontal transfers which will obscure the true phylogeny. In addition to the use of molecular data, the reconstitution of the phylogenetic tree must therefore also take into account morphological and fossil, biological and physiological characters. In particular, the structure of the flagellar apparatus appears to be a good phylogenetic marker.
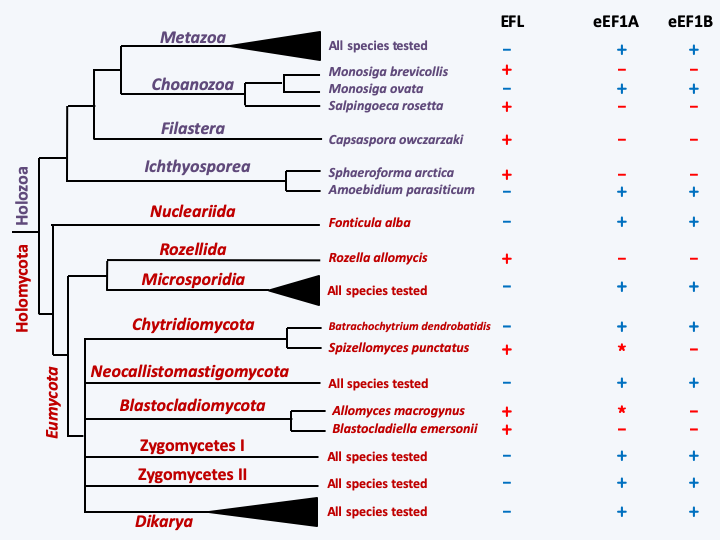
Figure 77.
Orthologs and paralogs. The comparisons of orthologs no. 1 (or no. 2) make it possible to measure the speciation event 1. On the other hand, the comparison of paralogs measures the duplication events in the ancestor (and not the speciation event 2). Errors can therefore occur if deletions have eliminated certain homologs.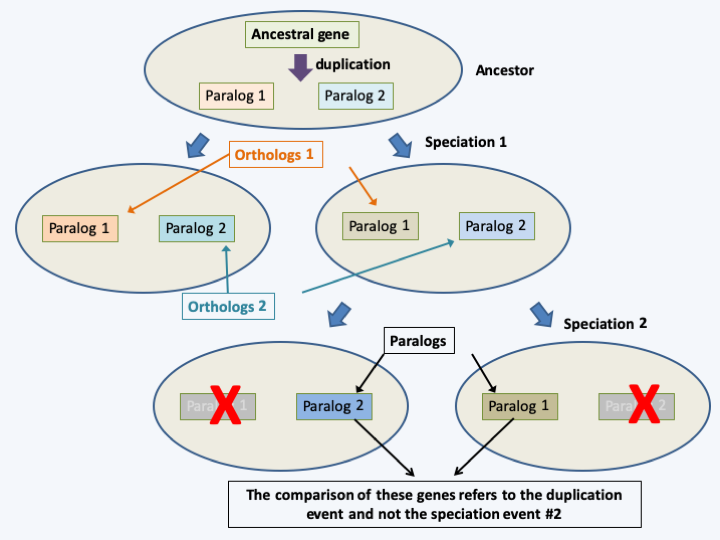
Figure 78.
Orthologs and paralogs. The comparisons of orthologs no. 1 (or no. 2) make it possible to measure the speciation event 1. On the other hand, the comparison of paralogs measures the duplication events in the ancestor (and not the speciation event 2). Errors can therefore occur if deletions have eliminated certain homologs.Among the successes of these phylogenies, in addition to the positioning within the eukaryotic tree of many groups whose origin was previously mysterious, it should be noted:
- Proof that eukaryotes derive from an organism that had mitochondria.
- Solving the complex stories of plastids and multicellularity.
- The determination that groups with surprising characters such as the absence of histone or different genetic codes are derived characters, that is, appeared late in evolution, and are not ancestral.
- Evidence that the diversity of microorganisms is much greater than that of animals and plants, which is understandable if these two groups derive from particular unicellular organisms.
- The confirmations that several groupings made on subtle morphological criteria do indeed correspond to monophyletic groups. For example, the Stramenopila put together on the criteria of the presence of two asymmetric flagella (see Figure 64) do indeed form a monophyletic group comprising organisms of the protozoan type, unicellular and multicellular algae, and fungal-like organisms (Figure 79).
- The demonstration that certain assemblages are polyphyletic or paraphyletic, including algae, and fungi, algae and protozoa.
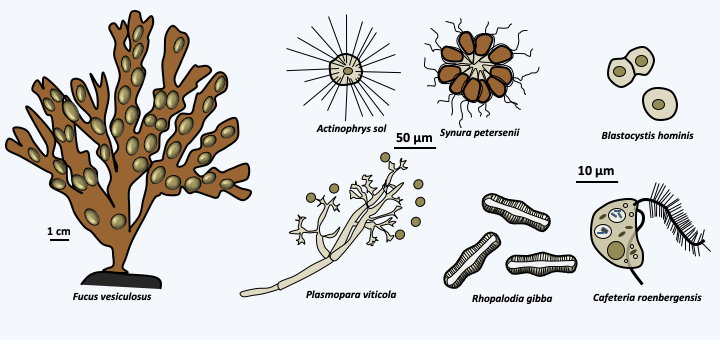
Figure 79.
Examples of Stramenopiles. Although heterogeneous in size, shapes and styles of life, these organisms are related and share the differentiation of asymmetric flagella. Note that in some species this synapomorphy has been lost! Indeed, some species have only one flagellum and others, such as the parasite Blastocystis, have lost it completely.Although progress is being made continuously, the main challenges of molecular methods, as explained above, lie in the difficulty in resolving the kinship relationships between great lineages, whether the eukaryotes themselves with the Eubacteria and the Archea, between the three major lineages of eukaryotes (Amorphea, Excavata and Diphoretickes) and virtually all important evolutionary radiations, including those of Metazoa, Streptophyta, Eumycota and Stramenopila.
Back to chapter index